Preprocess data and add prior GRN information using a human hindbrain dataset¶
In this notebook, we demonstrate the preprocessing steps and adding prior gene regulatory network (GRN) information needed before running the RegVelo pipeline. The dataset used in this tutorial is a subset of the first-trimester developing human brain dataset collected in Braun, E. et al, 2023.
A detailed description of the preprocessing steps is provided in the RegVelo manuscript.
Library import¶
import scanpy as sc
import numpy as np
import pandas as pd
import scvelo as scv
import scvi
import regvelo as rgv
General settings¶
scvi.settings.seed = 0
scv.settings.set_figure_params("scvelo", dpi=80, transparent=True, fontsize=14, color_map="viridis")
%matplotlib inline
Load data¶
In the following, we load the embryonic hindbrain dataset, that has already been annotated (see RegVelo manuscript). We further load the GRN learned from the human embryonic hindbrain single-cell multi-ome dataset (see RegVelo manuscript).
adata = rgv.datasets.hindbrain(data_type = "original")
adata
AnnData object with n_obs × n_vars = 49469 × 30958
obs: 'background_fraction', 'cell_probability', 'cell_size', 'droplet_efficiency', 'assignment', 'scDblFinder_DropletType', 'scDblFinder_Score', 'scrublet_DropletType', 'Tissue', 'batch', 'Experiment', 'Type', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'scrublet_score', 'scrublet_cluster_score_sample', 'scrublet_bh_pval_sample', 'background_fraction_cluster_score_sample', 'background_fraction_bh_pval_sample', 'paper_code', 'method', 'method2', 'FACS', 'stage', 'pcw_cont', 'bulk_name', '10X_run', 'n_genes', 'S_score', 'G2M_score', 'phase', 'leiden_res1', 'leiden_res1_R', 'Celltypist_DHB_predicted_labels', 'Celltypist_DHB_over_clustering', 'Celltypist_DHB_majority_voting', 'Celltypist_DHB_conf_score', 'Celltypist_GSE155121_full_predicted_labels', 'Celltypist_GSE155121_full_over_clustering', 'Celltypist_GSE155121_full_majority_voting', 'Celltypist_GSE155121_full_conf_score', 'Celltypist_GSE157329_developmental_system_full_predicted_labels', 'Celltypist_GSE157329_developmental_system_full_over_clustering', 'Celltypist_GSE157329_developmental_system_full_majority_voting', 'Celltypist_GSE157329_developmental_system_full_conf_score', 'Celltypist_GSE157329_annotation_full_predicted_labels', 'Celltypist_GSE157329_annotation_full_over_clustering', 'Celltypist_GSE157329_annotation_full_majority_voting', 'Celltypist_GSE157329_annotation_full_conf_score', 'Celltypist_GSE157329_final_annotation_full_predicted_labels', 'Celltypist_GSE157329_final_annotation_full_over_clustering', 'Celltypist_GSE157329_final_annotation_full_majority_voting', 'Celltypist_GSE157329_final_annotation_full_conf_score', 'STEMS_annotation_l1', '_scvi_batch', '_scvi_labels', 'leiden_SCVI', 'Celltypist_Immune_All_High_predicted_labels', 'Celltypist_Immune_All_High_over_clustering', 'Celltypist_Immune_All_High_majority_voting', 'Celltypist_Immune_All_High_conf_score', 'Celltypist_Immune_All_Low_predicted_labels', 'Celltypist_Immune_All_Low_over_clustering', 'Celltypist_Immune_All_Low_majority_voting', 'Celltypist_Immune_All_Low_conf_score', 'Teichmann_Celltype_fig1_full_predicted_labels', 'Teichmann_Celltype_fig1_full_over_clustering', 'Teichmann_Celltype_fig1_full_majority_voting', 'Teichmann_Celltype_fig1_full_conf_score', 'Teichmann_bone_full_predicted_labels', 'Teichmann_bone_full_over_clustering', 'Teichmann_bone_full_majority_voting', 'Teichmann_bone_full_conf_score', 'Teichmann_anatomical_site_full_predicted_labels', 'Teichmann_anatomical_site_full_over_clustering', 'Teichmann_anatomical_site_full_majority_voting', 'Teichmann_anatomical_site_full_conf_score', '_scvi_raw_norm_scaling', 'STEMS_annotation_l2', 'STEMS_annotation_l3', 'CellClass', 'CellCycleFraction', 'Clusters', 'Donor', 'DoubletFlag', 'DoubletScore', 'DropletClass', 'MitoFraction', 'NGenes', 'PrevClusters', 'Region', 'Sex', 'Subdivision', 'Subregion', 'TopLevelCluster', 'TotalUMIs', 'UnsplicedFraction', 'ValidCells', 'MB_Annotation_mb', 'MB_Clusters', 'MB_TopLevelCluster', 'vMB_Clusters', 'vMB_LRprediction_labels', 'CellClass_Subregion', 'MB_regvelo_annotation', 'vMB_regvelo_annotation', 'reference', 'STEMS_annotation_l2_SCANVI', 'STEMS_annotation_l2_prediction', 'MB_regvelo_annotation_SCANVI', 'MB_regvelo_annotation_prediction', 'vMB_regvelo_annotation_SCANVI', 'vMB_regvelo_annotation_prediction', 'CellClass_Subregion_SCANVI', 'CellClass_Subregion_prediction', 'leiden', 'batch_hvg', 'regvelo_annotation', 'regvelo_state'
var: 'gene_id'
uns: 'CellClass_Subregion_colors', 'Experiment_colors', 'MB_regvelo_annotation_colors', 'MB_regvelo_annotation_prediction_colors', 'STEMS_annotation_l2_colors', '_scvi_manager_uuid', '_scvi_uuid', 'batch_colors', 'hvg', 'leiden', 'leiden_SCVI', 'log1p', 'neighbors', 'pca', 'reference_colors', 'regvelo_annotation_colors', 'regvelo_state_colors', 'tsne', 'umap', 'vMB_regvelo_annotation_colors', 'vMB_regvelo_annotation_prediction_colors'
obsm: 'X_Embedding', 'X_Factors', 'X_mde_scanvi_CellClass_Subregion', 'X_mde_scanvi_MB_regvelo_annotation', 'X_mde_scanvi_STEMS_annotation_l2', 'X_mde_scanvi_vMB_regvelo_annotation', 'X_pca', 'X_scANVI_CellClass_Subregion', 'X_scANVI_MB_regvelo_annotation', 'X_scANVI_STEMS_annotation_l2', 'X_scANVI_vMB_regvelo_annotation', 'X_scVI', 'X_scVI_mde', 'X_tsne', 'X_umap', '_scvi_extra_categorical_covs', 'gene_expression_encoding'
layers: 'lognorm', 'spliced', 'unspliced'
obsp: 'connectivities', 'distances'
sc.pp.neighbors(adata, n_neighbors=30, n_pcs=50, use_rep="X_scVI")
sc.tl.umap(adata)
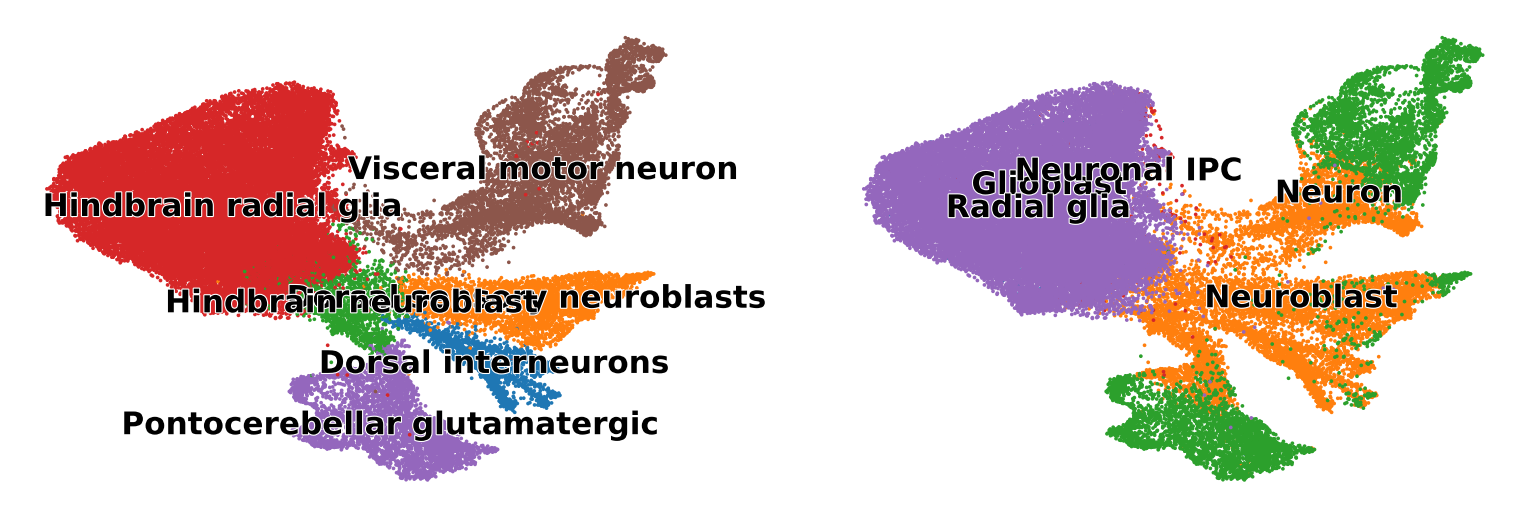
scv.pl.scatter(adata, basis="umap", title="", color=["regvelo_annotation", "CellClass"], legend_loc="on data")
eGRN = rgv.datasets.hindbrain_grn()
Create prior GRN for RegVelo¶
In the following, we preprocess the loaded GRN that will be needed as prior GRN for the RegVelo pipeline.
eGRN = eGRN.loc[:,["TF","Gene"]].copy()
reg = pd.crosstab(eGRN['TF'], eGRN['Gene'])
TF = np.unique(reg.index.tolist())
genes = np.unique(TF.tolist() + reg.columns.tolist())
GRN = pd.DataFrame(0, index=genes, columns=genes)
GRN.loc[TF,reg.columns.tolist()] = np.array(reg)
mask = (GRN.sum(0) != 0) | (GRN.sum(1) != 0)
GRN = GRN.loc[mask,mask].copy()
print("Done! processed GRN with " + str(reg.shape[0]) + " TFs and " + str(reg.shape[1]) + " targets")
Done! processed GRN with 151 TFs and 4219 targets
Preprocess data and align prior GRN for RegVelo pipeline¶
We perform preprocessing steps, consisting of filtering and normalization. We further compute the first and second order moments (means and uncentered variances) using scv.pp.moments needed for velocity estimation. Note that this step might be time-consuming.
Note
If preprocessing steps have already performed, you can skip this section and proceed directly to loading prior GRN.
scv.pp.filter_genes(adata, min_shared_counts=20)
scv.pp.normalize_per_cell(adata)
scv.pp.filter_genes_dispersion(adata, n_top_genes=3000)
scv.pp.moments(adata, n_pcs=None, n_neighbors=None)
Filtered out 19200 genes that are detected 20 counts (shared).
Normalized count data: X, spliced, unspliced.
Extracted 3000 highly variable genes.
computing moments based on connectivities
finished (0:00:06) --> added
'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
adata
AnnData object with n_obs × n_vars = 49469 × 3000
obs: 'background_fraction', 'cell_probability', 'cell_size', 'droplet_efficiency', 'assignment', 'scDblFinder_DropletType', 'scDblFinder_Score', 'scrublet_DropletType', 'Tissue', 'batch', 'Experiment', 'Type', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'scrublet_score', 'scrublet_cluster_score_sample', 'scrublet_bh_pval_sample', 'background_fraction_cluster_score_sample', 'background_fraction_bh_pval_sample', 'paper_code', 'method', 'method2', 'FACS', 'stage', 'pcw_cont', 'bulk_name', '10X_run', 'n_genes', 'S_score', 'G2M_score', 'phase', 'leiden_res1', 'leiden_res1_R', 'Celltypist_DHB_predicted_labels', 'Celltypist_DHB_over_clustering', 'Celltypist_DHB_majority_voting', 'Celltypist_DHB_conf_score', 'Celltypist_GSE155121_full_predicted_labels', 'Celltypist_GSE155121_full_over_clustering', 'Celltypist_GSE155121_full_majority_voting', 'Celltypist_GSE155121_full_conf_score', 'Celltypist_GSE157329_developmental_system_full_predicted_labels', 'Celltypist_GSE157329_developmental_system_full_over_clustering', 'Celltypist_GSE157329_developmental_system_full_majority_voting', 'Celltypist_GSE157329_developmental_system_full_conf_score', 'Celltypist_GSE157329_annotation_full_predicted_labels', 'Celltypist_GSE157329_annotation_full_over_clustering', 'Celltypist_GSE157329_annotation_full_majority_voting', 'Celltypist_GSE157329_annotation_full_conf_score', 'Celltypist_GSE157329_final_annotation_full_predicted_labels', 'Celltypist_GSE157329_final_annotation_full_over_clustering', 'Celltypist_GSE157329_final_annotation_full_majority_voting', 'Celltypist_GSE157329_final_annotation_full_conf_score', 'STEMS_annotation_l1', '_scvi_batch', '_scvi_labels', 'leiden_SCVI', 'Celltypist_Immune_All_High_predicted_labels', 'Celltypist_Immune_All_High_over_clustering', 'Celltypist_Immune_All_High_majority_voting', 'Celltypist_Immune_All_High_conf_score', 'Celltypist_Immune_All_Low_predicted_labels', 'Celltypist_Immune_All_Low_over_clustering', 'Celltypist_Immune_All_Low_majority_voting', 'Celltypist_Immune_All_Low_conf_score', 'Teichmann_Celltype_fig1_full_predicted_labels', 'Teichmann_Celltype_fig1_full_over_clustering', 'Teichmann_Celltype_fig1_full_majority_voting', 'Teichmann_Celltype_fig1_full_conf_score', 'Teichmann_bone_full_predicted_labels', 'Teichmann_bone_full_over_clustering', 'Teichmann_bone_full_majority_voting', 'Teichmann_bone_full_conf_score', 'Teichmann_anatomical_site_full_predicted_labels', 'Teichmann_anatomical_site_full_over_clustering', 'Teichmann_anatomical_site_full_majority_voting', 'Teichmann_anatomical_site_full_conf_score', '_scvi_raw_norm_scaling', 'STEMS_annotation_l2', 'STEMS_annotation_l3', 'CellClass', 'CellCycleFraction', 'Clusters', 'Donor', 'DoubletFlag', 'DoubletScore', 'DropletClass', 'MitoFraction', 'NGenes', 'PrevClusters', 'Region', 'Sex', 'Subdivision', 'Subregion', 'TopLevelCluster', 'TotalUMIs', 'UnsplicedFraction', 'ValidCells', 'MB_Annotation_mb', 'MB_Clusters', 'MB_TopLevelCluster', 'vMB_Clusters', 'vMB_LRprediction_labels', 'CellClass_Subregion', 'MB_regvelo_annotation', 'vMB_regvelo_annotation', 'reference', 'STEMS_annotation_l2_SCANVI', 'STEMS_annotation_l2_prediction', 'MB_regvelo_annotation_SCANVI', 'MB_regvelo_annotation_prediction', 'vMB_regvelo_annotation_SCANVI', 'vMB_regvelo_annotation_prediction', 'CellClass_Subregion_SCANVI', 'CellClass_Subregion_prediction', 'leiden', 'batch_hvg', 'regvelo_annotation', 'regvelo_state', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts'
var: 'gene_id', 'gene_count_corr', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'CellClass_Subregion_colors', 'Experiment_colors', 'MB_regvelo_annotation_colors', 'MB_regvelo_annotation_prediction_colors', 'STEMS_annotation_l2_colors', '_scvi_manager_uuid', '_scvi_uuid', 'batch_colors', 'hvg', 'leiden', 'leiden_SCVI', 'log1p', 'neighbors', 'pca', 'reference_colors', 'regvelo_annotation_colors', 'regvelo_state_colors', 'tsne', 'umap', 'vMB_regvelo_annotation_colors', 'vMB_regvelo_annotation_prediction_colors', 'CellClass_colors'
obsm: 'X_Embedding', 'X_Factors', 'X_mde_scanvi_CellClass_Subregion', 'X_mde_scanvi_MB_regvelo_annotation', 'X_mde_scanvi_STEMS_annotation_l2', 'X_mde_scanvi_vMB_regvelo_annotation', 'X_pca', 'X_scANVI_CellClass_Subregion', 'X_scANVI_MB_regvelo_annotation', 'X_scANVI_STEMS_annotation_l2', 'X_scANVI_vMB_regvelo_annotation', 'X_scVI', 'X_scVI_mde', 'X_tsne', 'X_umap', '_scvi_extra_categorical_covs', 'gene_expression_encoding'
layers: 'lognorm', 'spliced', 'unspliced', 'Ms', 'Mu'
obsp: 'connectivities', 'distances'
Note
The function rgv.pp.set_prior_grn aligns the loaded GRN with the gene expression data in adata and by default, it removes genes without incoming or outgoing regulatory edges.
adata = rgv.pp.set_prior_grn(adata, GRN.T)
adata
AnnData object with n_obs × n_vars = 49469 × 1273
obs: 'background_fraction', 'cell_probability', 'cell_size', 'droplet_efficiency', 'assignment', 'scDblFinder_DropletType', 'scDblFinder_Score', 'scrublet_DropletType', 'Tissue', 'batch', 'Experiment', 'Type', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'scrublet_score', 'scrublet_cluster_score_sample', 'scrublet_bh_pval_sample', 'background_fraction_cluster_score_sample', 'background_fraction_bh_pval_sample', 'paper_code', 'method', 'method2', 'FACS', 'stage', 'pcw_cont', 'bulk_name', '10X_run', 'n_genes', 'S_score', 'G2M_score', 'phase', 'leiden_res1', 'leiden_res1_R', 'Celltypist_DHB_predicted_labels', 'Celltypist_DHB_over_clustering', 'Celltypist_DHB_majority_voting', 'Celltypist_DHB_conf_score', 'Celltypist_GSE155121_full_predicted_labels', 'Celltypist_GSE155121_full_over_clustering', 'Celltypist_GSE155121_full_majority_voting', 'Celltypist_GSE155121_full_conf_score', 'Celltypist_GSE157329_developmental_system_full_predicted_labels', 'Celltypist_GSE157329_developmental_system_full_over_clustering', 'Celltypist_GSE157329_developmental_system_full_majority_voting', 'Celltypist_GSE157329_developmental_system_full_conf_score', 'Celltypist_GSE157329_annotation_full_predicted_labels', 'Celltypist_GSE157329_annotation_full_over_clustering', 'Celltypist_GSE157329_annotation_full_majority_voting', 'Celltypist_GSE157329_annotation_full_conf_score', 'Celltypist_GSE157329_final_annotation_full_predicted_labels', 'Celltypist_GSE157329_final_annotation_full_over_clustering', 'Celltypist_GSE157329_final_annotation_full_majority_voting', 'Celltypist_GSE157329_final_annotation_full_conf_score', 'STEMS_annotation_l1', '_scvi_batch', '_scvi_labels', 'leiden_SCVI', 'Celltypist_Immune_All_High_predicted_labels', 'Celltypist_Immune_All_High_over_clustering', 'Celltypist_Immune_All_High_majority_voting', 'Celltypist_Immune_All_High_conf_score', 'Celltypist_Immune_All_Low_predicted_labels', 'Celltypist_Immune_All_Low_over_clustering', 'Celltypist_Immune_All_Low_majority_voting', 'Celltypist_Immune_All_Low_conf_score', 'Teichmann_Celltype_fig1_full_predicted_labels', 'Teichmann_Celltype_fig1_full_over_clustering', 'Teichmann_Celltype_fig1_full_majority_voting', 'Teichmann_Celltype_fig1_full_conf_score', 'Teichmann_bone_full_predicted_labels', 'Teichmann_bone_full_over_clustering', 'Teichmann_bone_full_majority_voting', 'Teichmann_bone_full_conf_score', 'Teichmann_anatomical_site_full_predicted_labels', 'Teichmann_anatomical_site_full_over_clustering', 'Teichmann_anatomical_site_full_majority_voting', 'Teichmann_anatomical_site_full_conf_score', '_scvi_raw_norm_scaling', 'STEMS_annotation_l2', 'STEMS_annotation_l3', 'CellClass', 'CellCycleFraction', 'Clusters', 'Donor', 'DoubletFlag', 'DoubletScore', 'DropletClass', 'MitoFraction', 'NGenes', 'PrevClusters', 'Region', 'Sex', 'Subdivision', 'Subregion', 'TopLevelCluster', 'TotalUMIs', 'UnsplicedFraction', 'ValidCells', 'MB_Annotation_mb', 'MB_Clusters', 'MB_TopLevelCluster', 'vMB_Clusters', 'vMB_LRprediction_labels', 'CellClass_Subregion', 'MB_regvelo_annotation', 'vMB_regvelo_annotation', 'reference', 'STEMS_annotation_l2_SCANVI', 'STEMS_annotation_l2_prediction', 'MB_regvelo_annotation_SCANVI', 'MB_regvelo_annotation_prediction', 'vMB_regvelo_annotation_SCANVI', 'vMB_regvelo_annotation_prediction', 'CellClass_Subregion_SCANVI', 'CellClass_Subregion_prediction', 'leiden', 'batch_hvg', 'regvelo_annotation', 'regvelo_state', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts'
var: 'gene_id', 'gene_count_corr', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'CellClass_Subregion_colors', 'Experiment_colors', 'MB_regvelo_annotation_colors', 'MB_regvelo_annotation_prediction_colors', 'STEMS_annotation_l2_colors', '_scvi_manager_uuid', '_scvi_uuid', 'batch_colors', 'hvg', 'leiden', 'leiden_SCVI', 'log1p', 'neighbors', 'pca', 'reference_colors', 'regvelo_annotation_colors', 'regvelo_state_colors', 'tsne', 'umap', 'vMB_regvelo_annotation_colors', 'vMB_regvelo_annotation_prediction_colors', 'CellClass_colors', 'regulators', 'targets', 'skeleton', 'network'
obsm: 'X_Embedding', 'X_Factors', 'X_mde_scanvi_CellClass_Subregion', 'X_mde_scanvi_MB_regvelo_annotation', 'X_mde_scanvi_STEMS_annotation_l2', 'X_mde_scanvi_vMB_regvelo_annotation', 'X_pca', 'X_scANVI_CellClass_Subregion', 'X_scANVI_MB_regvelo_annotation', 'X_scANVI_STEMS_annotation_l2', 'X_scANVI_vMB_regvelo_annotation', 'X_scVI', 'X_scVI_mde', 'X_tsne', 'X_umap', '_scvi_extra_categorical_covs', 'gene_expression_encoding'
layers: 'lognorm', 'spliced', 'unspliced', 'Ms', 'Mu'
obsp: 'connectivities', 'distances'
Note
The following steps ensure that only velocity-informative genes and TF genes are considered and updates adata.uns["skeleton"] accordingly. The selection of velocity-informative genes is done using rgv.pp.preprocess_data, which in addition to min-max scaling of the spliced and unspliced layers, filters genes with non-negative fitted degradation rates \(\gamma\) and non-negative \(R^2\) values from scv.tl.velocity with mode=deterministic. The function rgv.pp.filter_genes further refines the GRN, such that each gene has at least one regulator. This step further reduces the number of genes considered.
velocity_genes = rgv.pp.preprocess_data(adata.copy()).var_names.tolist()
# select TFs that regulate at least one gene
TF = adata.var_names[adata.uns["skeleton"].sum(1) != 0]
var_mask = np.union1d(TF, velocity_genes)
adata = adata[:, var_mask].copy()
adata = rgv.pp.filter_genes(adata)
adata = rgv.pp.preprocess_data(adata, filter_on_r2=False)
adata.var["velocity_genes"] = adata.var_names.isin(velocity_genes)
adata.var["TF"] = adata.var_names.isin(TF)
adata
computing velocities
finished (0:00:01) --> added
'velocity', velocity vectors for each individual cell (adata.layers)
Number of genes: 684
Number of genes: 628
Number of genes: 623
AnnData object with n_obs × n_vars = 49469 × 623
obs: 'background_fraction', 'cell_probability', 'cell_size', 'droplet_efficiency', 'assignment', 'scDblFinder_DropletType', 'scDblFinder_Score', 'scrublet_DropletType', 'Tissue', 'batch', 'Experiment', 'Type', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'scrublet_score', 'scrublet_cluster_score_sample', 'scrublet_bh_pval_sample', 'background_fraction_cluster_score_sample', 'background_fraction_bh_pval_sample', 'paper_code', 'method', 'method2', 'FACS', 'stage', 'pcw_cont', 'bulk_name', '10X_run', 'n_genes', 'S_score', 'G2M_score', 'phase', 'leiden_res1', 'leiden_res1_R', 'Celltypist_DHB_predicted_labels', 'Celltypist_DHB_over_clustering', 'Celltypist_DHB_majority_voting', 'Celltypist_DHB_conf_score', 'Celltypist_GSE155121_full_predicted_labels', 'Celltypist_GSE155121_full_over_clustering', 'Celltypist_GSE155121_full_majority_voting', 'Celltypist_GSE155121_full_conf_score', 'Celltypist_GSE157329_developmental_system_full_predicted_labels', 'Celltypist_GSE157329_developmental_system_full_over_clustering', 'Celltypist_GSE157329_developmental_system_full_majority_voting', 'Celltypist_GSE157329_developmental_system_full_conf_score', 'Celltypist_GSE157329_annotation_full_predicted_labels', 'Celltypist_GSE157329_annotation_full_over_clustering', 'Celltypist_GSE157329_annotation_full_majority_voting', 'Celltypist_GSE157329_annotation_full_conf_score', 'Celltypist_GSE157329_final_annotation_full_predicted_labels', 'Celltypist_GSE157329_final_annotation_full_over_clustering', 'Celltypist_GSE157329_final_annotation_full_majority_voting', 'Celltypist_GSE157329_final_annotation_full_conf_score', 'STEMS_annotation_l1', '_scvi_batch', '_scvi_labels', 'leiden_SCVI', 'Celltypist_Immune_All_High_predicted_labels', 'Celltypist_Immune_All_High_over_clustering', 'Celltypist_Immune_All_High_majority_voting', 'Celltypist_Immune_All_High_conf_score', 'Celltypist_Immune_All_Low_predicted_labels', 'Celltypist_Immune_All_Low_over_clustering', 'Celltypist_Immune_All_Low_majority_voting', 'Celltypist_Immune_All_Low_conf_score', 'Teichmann_Celltype_fig1_full_predicted_labels', 'Teichmann_Celltype_fig1_full_over_clustering', 'Teichmann_Celltype_fig1_full_majority_voting', 'Teichmann_Celltype_fig1_full_conf_score', 'Teichmann_bone_full_predicted_labels', 'Teichmann_bone_full_over_clustering', 'Teichmann_bone_full_majority_voting', 'Teichmann_bone_full_conf_score', 'Teichmann_anatomical_site_full_predicted_labels', 'Teichmann_anatomical_site_full_over_clustering', 'Teichmann_anatomical_site_full_majority_voting', 'Teichmann_anatomical_site_full_conf_score', '_scvi_raw_norm_scaling', 'STEMS_annotation_l2', 'STEMS_annotation_l3', 'CellClass', 'CellCycleFraction', 'Clusters', 'Donor', 'DoubletFlag', 'DoubletScore', 'DropletClass', 'MitoFraction', 'NGenes', 'PrevClusters', 'Region', 'Sex', 'Subdivision', 'Subregion', 'TopLevelCluster', 'TotalUMIs', 'UnsplicedFraction', 'ValidCells', 'MB_Annotation_mb', 'MB_Clusters', 'MB_TopLevelCluster', 'vMB_Clusters', 'vMB_LRprediction_labels', 'CellClass_Subregion', 'MB_regvelo_annotation', 'vMB_regvelo_annotation', 'reference', 'STEMS_annotation_l2_SCANVI', 'STEMS_annotation_l2_prediction', 'MB_regvelo_annotation_SCANVI', 'MB_regvelo_annotation_prediction', 'vMB_regvelo_annotation_SCANVI', 'vMB_regvelo_annotation_prediction', 'CellClass_Subregion_SCANVI', 'CellClass_Subregion_prediction', 'leiden', 'batch_hvg', 'regvelo_annotation', 'regvelo_state', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts'
var: 'gene_id', 'gene_count_corr', 'means', 'dispersions', 'dispersions_norm', 'highly_variable', 'velocity_genes', 'TF'
uns: 'CellClass_Subregion_colors', 'Experiment_colors', 'MB_regvelo_annotation_colors', 'MB_regvelo_annotation_prediction_colors', 'STEMS_annotation_l2_colors', '_scvi_manager_uuid', '_scvi_uuid', 'batch_colors', 'hvg', 'leiden', 'leiden_SCVI', 'log1p', 'neighbors', 'pca', 'reference_colors', 'regvelo_annotation_colors', 'regvelo_state_colors', 'tsne', 'umap', 'vMB_regvelo_annotation_colors', 'vMB_regvelo_annotation_prediction_colors', 'CellClass_colors', 'regulators', 'targets', 'skeleton', 'network'
obsm: 'X_Embedding', 'X_Factors', 'X_mde_scanvi_CellClass_Subregion', 'X_mde_scanvi_MB_regvelo_annotation', 'X_mde_scanvi_STEMS_annotation_l2', 'X_mde_scanvi_vMB_regvelo_annotation', 'X_pca', 'X_scANVI_CellClass_Subregion', 'X_scANVI_MB_regvelo_annotation', 'X_scANVI_STEMS_annotation_l2', 'X_scANVI_vMB_regvelo_annotation', 'X_scVI', 'X_scVI_mde', 'X_tsne', 'X_umap', '_scvi_extra_categorical_covs', 'gene_expression_encoding'
layers: 'lognorm', 'spliced', 'unspliced', 'Ms', 'Mu'
obsp: 'connectivities', 'distances'
The data is now preprocessed and we can proceed to comparing different RegVelo model setups in the next tutorial!
Note
The preprocessed data can also be directly accessed via rgv.datasets.hindbrain(data_type = "preprocessed").